Protocols
Neuron Culture Solutions | Neuron Culturing | Transfecting Neurons with Lipofectamine 2000 | Preparation of Brain Membrane Fractions | Biotinylation Assay of Receptor Endocytosis with Cleavable Biotin Reagent | PSD Prep from High-Density Cultured Neurons | Large-batch Procedure for Preparing PDL-coated Coverslips
Neuron Culture Solutions
 Hanks Plus ( HBSS+)
Hanks Plus ( HBSS+)- HBSS (w/o Ca2+, Mg2+)
- + 10mM HEPES
- + 33.3mM Glucose
- + 5ug/ml Gentamicin (1:10,000 dilution of 50mg.ml Gentamicin)
Dissection Medium
- Hanks Plus (HBSS+)
- + 0.3% BSA
- + 12mM MgSO4
Culture Medium
- Neurobasal Medium (100ml)
- + B27 supplement (2ml)
- + GlutaMAX I (1ml)
- + 1ug/ml Gentamicin (2ul of 50mg/ml)
Digestion Solution
- NaHCO3 (4.2mM)
- HEPES (25mM)
- NaCl (137mM)
- KCl (5mM)
- Na2HP4 (7mM)
- Adjust to pH 7.4
FUDR:
- 5-FLUORO-2?DEOXYURIDINE (F-0503 100mg SIGMA) 10mM 25mg
- URIDINE (U-3003 5g SIGMA) 10mM 24mg
- H2O
- Final Volume: 10ml
- Sterile filtration, 0.2um
AraC: Cytosine ?D-Arabino-Furanoside
back to top
Neuron Culturing
Coating (day before dissection)
Poly-D-lysine
1mg/ml 80ul/glass coverslip (or enough to cover the coverslip) 0.1mg/ml
2ml/60mm dish
Incubate at 37 or RT, O/N (or can go as short as 2 hrs if in a hurry)
(Day of dissection)
Wash coverslip or dish with water 2X
Laminin
1ug/ml 2ml/60mm dish (after dishes were coated with PDL)
Incubate at least 2h
Wash dishes with HBSS 2X.
Dish and plate can be filled with plating medium, keep in incubator
Dissection
Prepare ice-cold Dissection Medium (DM) w/CaCl2 (2mM) in dish and two 15ml-tube
All instruments in 70% Alcohol
4-6 Rats
Oval anesthesia: Halothane
1. Cut head right behind ears (large scissors and forceps with teeth) into dish cover
2. Cut skin and skull through the middle line, two cuts behind eyes (10cm scissors)
3. Lift skin and skull to expose brain (10cm graefe forceps)

4. Move brain into dish w/DM (Micro-spatula, a little bit curved)
5. ( 8cm angled-up spring scissors and 11cm fine shanks forceps)

6. Tear off the vessel membrane outside the cortex (11cm fine shanks forceps and 45 tip forceps)
7. Expose hippocampus (11cm fine shanks forceps and 45 tip forceps)
8. Cut hippocampus along outside line and roll it off ( 11cm fine shanks forceps and 8cm spring scissors)
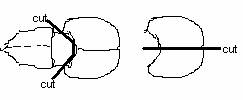
9. Collect all the cortex to a dish w/DM, chop them into pieces and pipette them in 15ml tube w/DM.
10. Roll and open hippocampus and cut correct region of hippocampus (11cm fine shanks forceps and scalpel), collect and chop them into pieces, pipette them in 15ml tube w/DM
Digestion
1. Spin down for 1min. 1000rpm.
2. *Rinse with Hanks Plus 10ml/tube
3. *Spin down for 1min. 1000rpm.
4. Dissolve 70mg Trypsin and 2mg DNAse in 8ml pre-warmed (37 ) Digestion Solution. Filter sterilize the digestion solution (DS).
5. Aspirate the Hanks Plus and incubate cortex pieces in 5ml sterile DS and hippocampus pieces in 3ml sterile DS, 5-8min. 37 agitate slowly from time to time.
6. Spin down for 1min. 1000rpm. And aspirate the supernatant.
7. During incubation, dissolve 40mg trypsin inhibitor in 8ml pre-warmed (37 ) Dissection Medium (DM). Filter sterilize to new tube.
8. Incubate hippocampus pieces with 3ml DM with trypsin inhibitor (5ml for cortical pieces) for 2min at 37 .
9. Spin down for 1min. 1000rpm. And aspirate the supernatant.
10. Wash with 10ml/tube ice cold DM. Spin down for 1min. 1000rpm. And aspirate the supernatant.
11. Dissolve 1mg DNAse in 5ml ice cold DM, filter sterilize. Add 1ml to hippocampus pieces and 3-4ml to cortex pieces.
12. Triturate slices very gentle. Avoid air bubbles (osmotic shock). First, use a fire polished glass Pasteur pipette, then use a fire polished pipette with reduced diameter
13. *Filter cells through the cell strainer (40um Falcon 2340), rinse with some DM.
Cell recovery
1. Spin down for 10min. 1000rpm. And aspirate the supernatant.
2. Re-suspend the hippocampus pellet in 5ml ice-cold DM or Hank's Plus and the cortex pellet in 5-10ml ice-cold DM or Hank's Plus.
Counting
10ul cells + 90ul Trypan Blue Stain 0.4% (15250-061 GIBCO)
Pipette 10ul of mixture above on Counting Chamber (15170-208 VWR)
The cell number inside 4x4 square will be N,
N x 105cell/ml --------cell concentration
Plating
Plate 2 million (106) cortical cells on each dish with Plating Medium.
Plate high or low density of hippocampal cell on each coverslip with Plating Medium.
Feeding
(3 days after culturing) Add FUDR (10uM) to cortical neurons.
(wait for perfect time - Glial cell layer reaches around 70% confluence) Add FUDR (10uM) to hippocampal cells
(twice a week) Replace half of the medium with fresh Culture Medium w/FUDR (10uM) for hippocampal neurons.
(twice a week) Replace half of the medium with fresh Culture Medium for cortical cells.
*: Optional steps that can be skipped. We usually skip them.
back to top
Transfecting Neurons with Lipofectamine 2000
To transfect cells in one well of a 12-well plate (~1ml growth medium in the well): 1) Mix 25µl Opti-MEM with 1µl Lipofectamine 2000. Let sit for 5 min at room temperature.
2) Mix 25µl Opti-MEM with 1 to 1.5µg cDNA of choice (for co-transfections, use 1 to 1.5µg of each construct). This does not need to sit. It is possible to use much less DNA.
3) Combine solutions from 1 and 2, and pipette gently to mix. Let sit for 20 min at room temperature.
4) Add total solution from 3 to well. Swirl to mix in the well.
Notes:
I never wash off the growth medium, except if I've transfected freshly plated neurons.
Using Opti-MEM is critical when pre-mixing the L2K. The neurons can be growing in Neurobasal and serum, though, when the mixture is added.
Transfection rates are quite high during the first week after plating (<10%) but very low after that. Expect only a few neurons per well to be transfected after a couple of weeks.
Dense cultures transfect much, much better than sparse ones, perhaps due to the presence of glia.
Expression starts to be visible in ~2-4 hours, and peaks after 24 hours.
Duration of expression is totally dependent on the construct.
Cell lines can be transfected efficiently with L2K as well, using 1/10th or less of the lipid.
back to top
Preparation of Brain Membrane Fractions
Crude Membrane/Cytosol Prep Be sure that all procedures are done with precooled reagents at 4°C. Dissect out brain regions of interest into ice-cold into 10 volumes of cold homogenization buffer (0.32 M sucrose, 10 mM HEPES pH 7.4, 2 mM EDTA, protease inhibitors, phosphatase inhibitors as below).
Homogenize using 10-15 strokes of a motor-driven glass-teflon homogenizer. Never use polytron.
Spin at 1000 x g for 15 min to remove pelleted nuclear fraction (P1).
Take supernatant (S1) and spin at ~200,000 x g (50,000 rpm for 30 min in 70.1 Ti rotor; 62,000 rpm for 15 min in TLA100.3 rotor) to yield crude cytosol (S2) and crude membrane pellet (P2).
Resuspend pellet in homogenization buffer.
Spin again at ~200,000 x g to yield washed crude membrane pellet (P2?.
Resuspend pellet in HEPES-Lysis buffer (50 mM HEPES pH 7.4, 2 mM EDTA, protease/phosphatase inhibitors).
Measure protein concentration by BCA or Coomassie.
Can solubilize with detergents or store at ?0°C.
Microsome Prep
Microsomes = vesicles derived from rough ER and various smooth membrane bound organelles (including Golgi stack components).
Homogenize brain in homogenization buffer (0.32 M sucrose, 10 mM HEPES pH 7.4, 2 mM EDTA, protease inhibitors, phosphatase inhibitors as below).
Spin at 1000 x g for 15 min to remove pelleted nuclear fraction (P1).
Take supernatant (S1) and spin at 10,000 x g for 20 min to remove pelleted mitochondria (P2).
Spin supernatant again at 12,000 x g for 30 min to doubly remove pelleted mitochondria (P2).
Take resulting S2 supernatant and centrifuge at 140,000 x g for 120 min to yield microsomal pellet (P3).
The resulting microsome pellet can be resuspended in homogenization buffer and further fractionated by layering over a discontinuous sucrose gradient (0.8, 1.0, 1.3, and 2.0 M sucrose) and centrifuging at 97,000 x g for 120 min.
I can’t quite remember what organelles are present at each interface, but Wenthold typically uses the interfaces between 1.0 and 0.8 M and between 0.8 M and 0.32 M.
For reference, see Gurd et al., (1974). J. Neurochem. 22:281-290.
Synaptic Plasma Membrane Prep
This protocol is adapted from Blackstone et al. (1992) and Lau et al. (1996) and should be followed when an enriched membrane prep or PSD fraction is desired
All procedures should be done at 4°C using precooled reagents. For rat and mouse samples, immediately remove brain from the cranium into ice cold HEPES-buffered sucrose (0.32 M sucrose, 4 mM HEPES pH 7.4) containing a freshly added protease inhibitor cocktail (required, see below) and phosphatase inhibitor cocktail (optional, see below). Different parts of the brain can be subdissected and enriched plasma membranes/PSD can be prepared as described below.